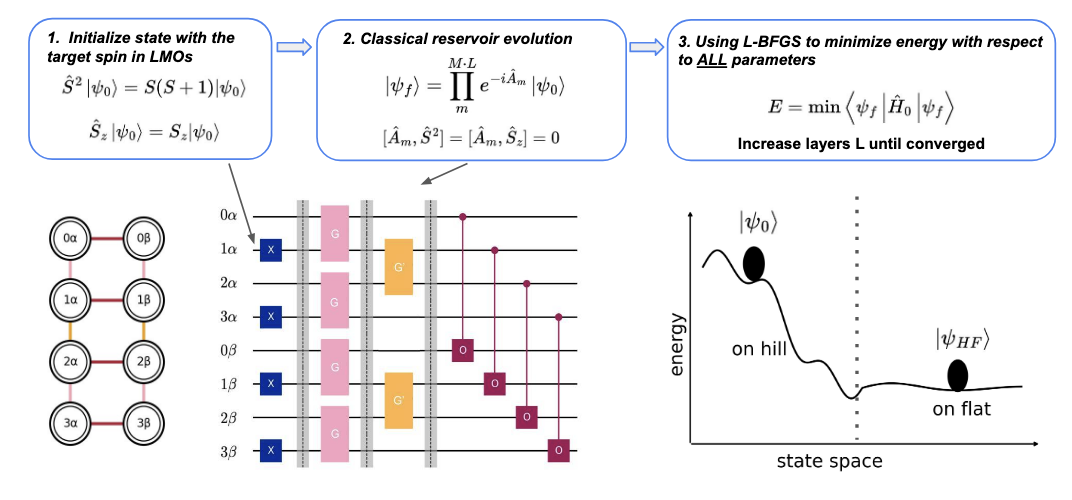
Abstract
Ground state preparation is a central application of quantum algorithms for electronic structure. We introduce the classical reservoir approach, a low cost variational ansatz tailored to near-term hardware, requiring only nearest-neighbor interactions on a machine with square-lattice connectivity. Unlike traditional methods built from the classically efficient Hartree Fock theory, our ansatz operates in localized molecular orbitals to study previously unexplored regions of the variational parameter space. Numerical benchmarks demonstrate chemical accuracy across diverse systems and bond lengths; notably, significantly reduced circuit depths are attainable when relaxed error thresholds (e.g., tens of Eh) are permissible. We benchmark the method on hydrogen chains, N2, O2, CO, BeH2, and H2O, the latter corresponding to an effective 24 qubit calculation.